
Here, Professor Kazunari Domen, Shinshu University and The University of Tokyo, explains a particulate photocatalyst producing hydrogen from water by perfect photon-to-chemical conversion, which is essential to meet our future need for renewable hydrogen
Water splitting reaction driven by solar energy is a technology for producing renewable solar hydrogen on a large scale. To put such technology to practical use, the production cost of solar hydrogen must be significantly reduced [1]. This requires the reaction system that can split water efficiently and can be scaled up easily. A system consisting of particulate semiconductor photocatalysts can be expanded over a large area with relatively simple processes. Therefore, it will make great strides toward large-scale solar hydrogen production if photocatalysts driving the sunlight-driven water splitting reaction with high efficiency are developed.
To upgrade the solar energy conversion efficiency of photocatalytic water splitting, it is necessary to improve two factors: widening the wavelength range of light used by the photocatalyst for the reaction and increasing the quantum yield at each wavelength. The former is determined by the bandgap of the photocatalyst material used, and the latter is determined by the quality of the photocatalyst material and the functionality of the cocatalyst used to promote the reaction. However, photocatalytic water splitting is an endergonic reaction involving multi-electron transfer occurring in a non-equilibrium state.
Therefore, there are many opportunities for recombination of photoexcited electrons and holes as well as backward reactions, and so the quantum yield tends to decrease. In fact, only a few photocatalysts developed so far split water with quantum yields of 50% or higher, and most of them exhibit quantum yields of 10% or less even for wide-bandgap oxides that require ultraviolet light excitation. This raises an important and essential question as to whether the quantum yield inevitably decreases in the photocatalytic water splitting reaction or whether proper structural and functional design is missing. To answer this ultimate question, Prof Domen’s group studied the material preparation and cocatalyst loading methods for strontium titanate (SrTiO3), a traditional ultraviolet light-responsive water-splitting photocatalyst, and finally revealed that photocatalytic water splitting with a quantum yield close to unity can be realised [2].
The path to the near-perfect reaction
SrTiO3 powder is prepared by a flux method allowing the growth of highly crystalline particles. However, particles tend to grow to a micrometre size, which is too large for photocatalytic applications. In addition, the material readily contains oxygen vacancies because the oxide is in equilibrium with the oxygen in the atmosphere. As a consequence, tetravalent titanium ions are partly reduced, which facilitates charge recombination. However, the particle growth can be suppressed by adding aluminium oxide during synthesis by the flux method [3]. Also, the trivalent aluminium ions replace tetravalent titanium ions in SrTiO3, and the oxide material becomes charge-neutral in the presence of oxygen vacancies. This intentional introduction of oxygen vacancies effectively suppresses the generation of additional oxygen defects and the associated reduction of tetravalent titanium species [4].

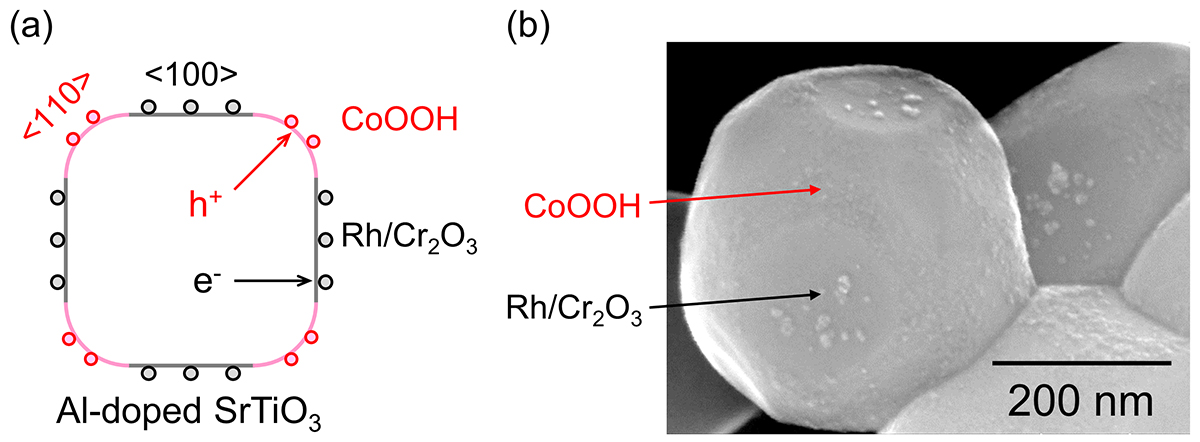
The Al-doped SrTiO3 particles have a truncated cube shape that exposes the {100} and {110} planes, reflecting the perovskite-type structure. In such semiconductor fine particles, photoexcited electrons and holes can be rectified in different directions and spatially separated because of an anisotropic potential gradient inside the particles [2]. Specifically, photoexcited electrons and holes selectively migrate to the <100> and <110> directions, respectively (Fig. 1a). This anisotropic charge transfer can be utilised for site-selective coloading of cocatalysts by photodeposition. In the photodeposition method, the hydrogen evolution cocatalyst (namely, core/shell-structured Rh/Cr2O3) is deposited on the facets where the photoexcited electrons reach by the reduction reaction, [5] and the oxygen evolution cocatalyst (namely, CoOOH) is deposited on the other facets where photoexcited holes reach by the oxidation reaction [6]. In the electron microscope image (Fig. 1b), fine particles of the hydrogen evolution cocatalyst and the oxygen evolution cocatalyst are respectively seen on the {100} and {110} planes [2]. In this way, the reduction sites and the oxidation sites are spatially separated, and thus charge carriers are separated efficiently.
Furthermore, the consumption of electrons and holes in the water splitting reaction is promoted by the supported cocatalysts. As a result, recombination is almost completely suppressed, and the water splitting reaction with a quantum yield close to unity is achieved. Here, the Rh/Cr2O3 co-catalyst demonstrates excellent reaction characteristics in that it shows high activity in the water reduction reaction while it does not catalyse the oxygen reduction reaction or the water formation reaction from hydrogen and oxygen [7]. The use of such highly active and selective cocatalysts is also important in preventing the decrease in quantum yield.
Future of the photocatalyst technology
The suppression of particle growth and titanium reduction of the SrTiO3 photocatalyst and the site-selective coloading of highly active and selective cocatalysts allowed for efficient spatial separation of photoexcited electrons and holes and their rapid consumption by forward reactions. As a result, the photocatalytic water splitting reaction proceeded with a quantum yield of almost unity. This achievement means that a particulate photocatalyst with a relatively simple structure can split water with the same high quantum yield as the photochemical conversion process in natural photosynthesis relying on complex protein structures. Therefore, it has great significance in the design and analysis of particulate photocatalysts for water splitting.
It is estimated that a solar energy conversion efficiency of 5 to 10% is required for the practical application of solar hydrogen production via photocatalytic water splitting [1]. The Al-doped SrTiO3 used this time has a large bandgap and can only use ultraviolet light. Therefore, it is not practical for solar energy conversion. In the future, it will be necessary to improve the quantum yield of photocatalysts that have narrow bandgaps and can use light in a wide range of visible light for the effective harvesting of solar energy. It is known that certain oxynitrides, nitrides, and oxychalcogenides have band structures suitable for water splitting under visible light irradiation (Fig. 2). Advanced photocatalyst preparation technology is required to decompose water using such non-oxide particles. However, by aiming for the ideal structure of the photocatalyst and cocatalyst revealed this time, it will be possible to develop visible-light-responsive photocatalysts for highly efficient sunlight-driven water splitting in the future. Once particulate photocatalysts highly active in the sunlight-driven water splitting reaction are developed, such photocatalysts will be processed into panel reactors and expanded over a large area. The development of such water splitting and hydrogen production plants is also an important issue toward practice application of this technology and is currently under investigation.
References
[1] Hisatomi et al., Nature Catal. 2, 387 (2019). https://www.nature.com/articles/s41929-019-0242-6
[2] Takata et al., Nature 581, 411–414 (2020). https://www.nature.com/articles/s41586-020-2278-9
[3] Ham et al., J. Mater. Chem. A, 4, 3027 (2016). https://pubs.rsc.org/en/content/articlelanding/2016/ta/c5ta04843e
[4] Takata et al., J. Phys. Chem. C, 113, 19386 (2009). https://pubs.acs.org/doi/10.1021/jp908621e
[5] Maeda et al., Angew. Chem. Int. Ed., 45, 7806 (2006). https://onlinelibrary.wiley.com/doi/full/10.1002/anie.200602473
[6] Lyu et al., Chem. Sci., 10, 3196 (2019). https://pubs.rsc.org/en/content/articlelanding/2019/sc/c8sc05757e
[7] Yoshida et al., J. Phys. Chem. C, 113, 1015 (2009). https://pubs.acs.org/doi/abs/10.1021/jp901418u
Please note: This is a commercial profile








