
Here, we find out about first-principles computer simulations performed in the group of Prof Mark E. Tuckerman, who uncover principles for designing high-performance alkaline anion exchange membrane (AEM) fuel cells
Climate change is an issue we have all become increasingly aware of over the last decade. It is known that its main cause is the production of greenhouse gases, such as carbon dioxide from the burning of fossil fuels.
Many research groups are working to develop renewable energy technologies that will reduce the need to burn fossil fuels and ultimately, eliminate our reliance on them altogether. Currently, considerable attention is focused on electrochemical energy sources such as batteries and fuel cells. Our team are employing techniques of mathematical modelling and computer simulation to design new realisations of these technologies. This article will briefly describe our work in the area of alkaline anion exchange membrane (AEM) fuel cells.
Why AEMs?
Generally, fuel cells are electrochemical devices capable of delivering power by converting the chemical energy stored in a fuel, such as hydrogen, into electricity. When hydrogen fuel is used, the only waste products are water – which can be recycled back into the fuel cell – and heat. Fuel cells can be used to power anything that runs on electricity, making them ideal for common devices, such as computers and mobile phones. They can, additionally, be used to power vehicles, where efficiencies can reach 40-60% compared to the approximately 20% efficiency of typical petrol engines.
Commonly used proton-exchange membrane (or PEM) fuel cells operate under acidic conditions, employing electrochemical reactions that produce protons at the anode, which are consumed at the cathode. Transporting the protons from anode to cathode using an electrolyte material – the PEM – consisting of an acidic polymer dissolved in water. Breaking down the fuel requires a precious-metal catalyst, such as platinum, that can withstand the corrosive effect of the acidic environment.
Although AEM fuel cells bear some similarity to PEM fuel cells, their primary advantage is that they operate under alkaline or basic conditions for which non-precious metal catalysts can be employed, thus making them better for mass production.
At the cathode of an AEM fuel cell, oxygen is combined with water and electrons to produce hydroxide ions. When fed with hydrogen fuel, the hydrogen molecules at the anode are combined with the hydroxide ions produced in the cathode reaction to produce water and electrons. The role of the AEM is to transport hydroxide ions from the cathode to the anode. Analogous to PEMs, AEMs generally consist of a basic polymer dissolved in water. However, designing polymeric materials capable of achieving the high rate of hydroxide transport necessary for efficient fuel-cell operation constitutes a significant challenge in polymer engineering and synthesis that can benefit significantly by leveraging the mathematical methods of theoretical chemistry and computational modelling.
The role and impact of computational modelling
The transport of protons and hydroxide ions through water does not occur via simple hydrodynamic diffusion but rather through a chemical process whose mechanism was first revealed by first-principles computer simulation1,2. The approach employed in these studies involves numerically solving the fundamental equations of mechanics (F = ma or Newton’s 2nd law) for the motion of every atom in a system, with interatomic forces (F) obtained from electronic structure calculations that are performed “on the fly” at each step of the simulation.
These simulations reveal that transport of aqueous protons and hydroxide ions, which should be viewed as local topological defects in water’s complex and dynamic network of hydrogen bonds, occurs via specific rearrangements of the hydrogen bonds between the ions and surrounding water molecules that “activate” the local environment to promote a proton transfer reaction between the ion and a nearby water molecule. This reaction moves the core of the topological defect from one point to another in the hydrogen bond network. In this process, it is not a single ion that diffuses but rather a core of positive or negative charge that hops from site to site in the liquid through the interconversion of water molecules into ions via sequential proton-transfer reactions.
AEMs generally consist of a polymer backbone functionalised by cation groups that, when the AEM is hydrated, dangle into the aqueous region3. Laboriously synthesising and testing, in actual fuel cells, all the possible membrane systems that can be made would be an intractable feat. Computational modelling can inform synthetic efforts by rapidly exploring the parameter space and revealing key design features.
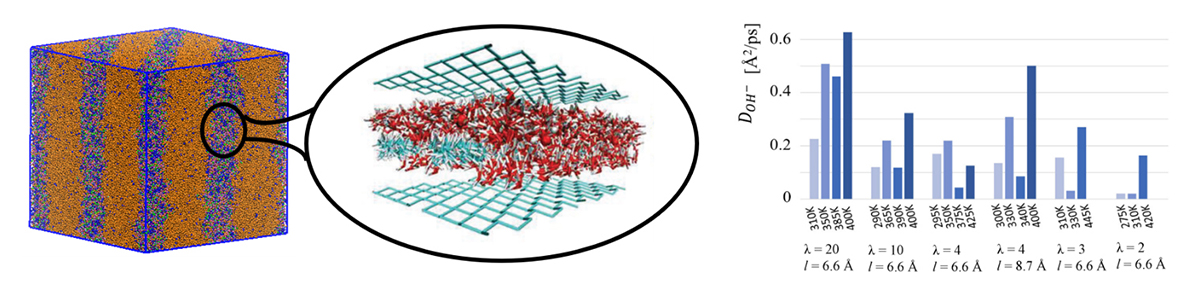
Our computational model consists of an idealised confined environment that mimics the ion diffusion channels revealed by coarse-grained simulation4 (Figure 1). Within our model, a large number of design parameters are being explored, such as the chemical composition of the polymer, placement of the positively charged groups, chemical structure of the positively charged groups, and optimal water content.
Our simulations indicate that two key factors in AEMs are the water distribution within the membrane and water mobility around the cations5,6. All other design parameters should be chosen to produce morphologies that promote such optimal water distributions and hydrogen-bond topologies; tuning the temperature proves instrumental in influencing the mobility. Importantly, we have identified design strategies for producing favourable water distributions at both high and low levels of hydration.
Next steps
The simulation studies we have carried out, thus far, have already identified several candidate materials. Nevertheless, we will continue to explore more AEM prototypes. A long-term goal of our work will be to arrive at a general set of design principles which, if applied, could lead to AEMs of a greater efficiency and, ultimately, to the creation of a class of new, clean electrochemical power sources.
References
1 M. E. Tuckerman, D. Marx, M. Parrinello, The nature and transport mechanism of hydrated hydroxide ions in aqueous solution, Nature, 417, 925-929, 2002.
2 D. Marx, A. Chandra, M. E. Tuckerman, Aqueous basic solutions: Hydroxide solvation, structural diffusion, and comparison to the hydrated proton, Chem. Rev. 110, 2174-2216, 2010.
3 J. R. Varcoe, et al. Anion-exchange membranes in electrochemical systems. Energy & Environ. Sci. 7, 3135-3191, 2014.
4 F. Sepehr, et al. Mesoscale simulations of anion exchange
membranes based on quaternary ammonium tethered triblock copolymers. Macromol. 50, 4397-4405, 2017.
5 T. Zelovich, et al. Ab initio molecular dynamics study of hydroxide diffusion mechanisms in nanoconfined structural mimics of anion exchange membranes. J. Phys. Chem. C 128, 4639-4653, 2019.
6 T. Zelovich, et al. Hydroxide ion diffusion in anion-exchange
membranes at low hydration: Insights from ab initio molecular dynamics. Chem. Mater. 31, 5778-5787, 2019.
*Please note: This is a commercial profile








