Dr Charlie Carter from the University of North Carolina at Chapel Hill explores how advances in enzymology and phylogenetics enable biochemical measurements that could map the ancestral development of genetic coding
Chemical reactions in the cell degrade energy-rich food molecules and rearrange their chemical bonds into other molecules necessary to construct and sustain living things far from chemical equilibrium. Those reactions occur spontaneously at rates that span an extraordinary range, the fastest going up to 1025 times faster than the slowest. (1) All these reactions must be accelerated by different amounts to maintain adequate concentrations of chemical components in the cell. Enzymes perform that differential acceleration exquisitely well.
Enzymology and phylogenetics
Enzymes are the nanomachines that transform random chemistry into regulated metabolic pathways that sustain life. Their precisely shaped cavities, called active sites, can tell one molecule from another by how well they fit within the site. That unique talent enables enzymes to speed reactions up by the astronomical amounts required to synchronise life’s chemistry. It also provides the specificity necessary to build metabolic pathway networks.
Enzymes are proteins that the cell assembles according to genetic blueprints (genes). Reading those blueprints is the main information flow in living cells. Molecular evolution is the study of how enzymes assumed their present form. Phylognetic studies find successively more distant relatives and use those comparisons to construct family trees. Those trees, in turn, suggest the amino acid sequences of common ancestors. The ample gene sequence databases open access to the recent evolutionary history of enzymes and related regulatory proteins. (2-4)
The remote origins of the proteome, however, pose a greater challenge. Three-dimensional structures must guide the phylogenetic approach. Such analyses reveal that contemporary enzymes have a hierarchical modularity. Evolutionarily related enzymes use the same active sites to speed similar chemical reactions up. Nature then embeds these active sites, like Russian matryoshka dolls, into successive ‘layers’ of supplemental polypeptide that enhance function in mature enzymes and differentiate one family from another. That hierarchy points to the deeper roots of modern enzymes.
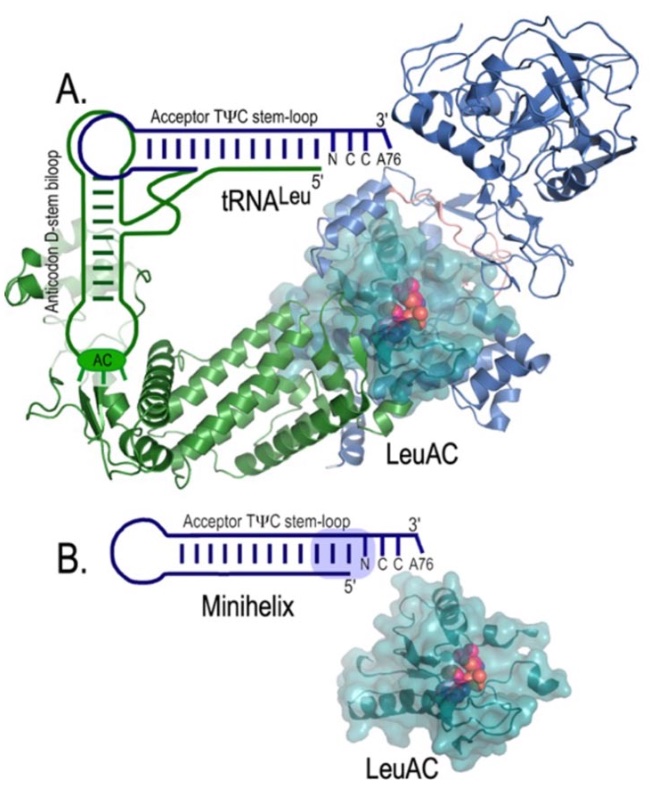
A. represents the contemporary leucyl-tRNA synthetase•tRNALeu cognate pair. LeuRS and tRNALeu have two domains: a catalytic domain that interacts with the tRNA acceptor stem and another domain which interacts with other parts of the tRNA. B. shows a functional single-domain system. The leucyl-tRNA synthetase urzyme, LeuAC, catalyses acylation of the tRNALeu minihelix, facilitating experimental measurement of the spectrum of functional amino acid and minihelix substrates, as well as the impact of the three base pairs (light blue shading) that determine cognate recognition.
Urzymology
A major step forward was deconstructing that modular hierarchy and expressing just the active site module from the two families (Class I and II) of aminoacyl-tRNA synthetases (AARS). (5-7) AARS are the nanomachines that actually translate the genetic code. They are central to how information is read from genes. To our surprise, these small excerpts require minimal re-design to behave like the mature contemporary enzymes. They are just weaker and less discriminating. We call them ‘urzymes’, contracting the prefix ‘ur’ (= ‘first’) with ‘enzyme’. Their amino acid sequences are also closely related, and their catalytic activities and substrate specificities resemble their full-length counterparts. These two properties make urzymes useful experimental models for the primordial emergence and early evolution of enzymes. (8)
Catalysis by full-length Class I AARS depends on two different active-site signature sequences, HIGH near the N- terminus and KMSKS at the C-terminus of the catalytic domain. Mutating the polar histidine and lysine residues in these signatures separately and in combination with both full-length leucyl-tRNA synthetase and its urzyme, LeuAC (9) allowed measurement of the energetic coupling energy coordinating the impact of the two signatures on catalysis. Remarkably, the two signatures function synergistically in the full-length LeuRS but oppose each other’s effects in urzymes. That result is prima facie evidence both that urzyme catalysis is real and that the domains eliminated to make the urzyme impose the coordinated behaviour of these signatures in the mature enzyme.
Urzymes enable experimental studies of how genetic coding began
It was never obvious how to investigate the molecular ancestry of genetic coding experimentally. Recent developments in urzymology now make that possible (see Fig. 1). While we were developing AARS urzymes, other groups found that full-length AARS could acylate the acceptor-stem domains of their cognate tRNAs.
A pivotal step in developing an experimental system came when we recently showed that AARS urzymes also acylate tRNA minihelices. (10) Generation of additional such pairs could be a game-changer by facilitating experimental measurements of the ranges of amino acid and tRNA minihelix substrates that can be aminoacylated by different urzyme•minihelix cognate pairs.
My previous piece (11) noted that AARS•tRNA cognate pairs are the computational AND gates that make genetic coding work. The specific coding properties of ancestral urzyme•minihelix cognate pairs Fig. 1B can help us understand what catalysts can be made using fewer coding letters. Experimental access to that novel type of information can help us map the growth of the coding alphabet.

This work is licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International.