With current treatment options for BTHS cardiomyopathic phenotype focused predominantly on alleviating symptoms, Dr Hani N. Sabbah, Director of Cardiovascular Research at Henry Ford Health, explores the potential of more targeted treatment approaches
Barth syndrome (BTHS) is a rare, X-linked disease caused by defects in the TAFAZZIN gene encoding an acyltransferase responsible for the remodeling/maturation of cardiolipin (critical to mitochondrial structure/function).(1) The most common clinical manifestation of BTHS is cardiomyopathy (~90% of BTHS patients) with a wide range of phenotypical presentations.(2,3) Mechanisms contributing to BTHS cardiomyopathy pathophysiology include abnormal mitochondrial structure/function,(4) defective mitochondrial calcium uptake,(5) a mismatch between ATP supply and demand,(6) and altered lipid/glucose metabolism.(7) The BTHS cardiomyopathic phenotypes appear to evolve with advancing age, which may be useful in treatment selection.
Natural history of BTHS cardiomyopathy
BTHS cardiomyopathic phenotypes include dilated cardiomyopathy (DCM), left ventricular (LV) noncompaction (LVNC), endocardial fibroelastosis, and hypertrophic cardiomyopathy (HCM). Presenting in infancy, DCM is characterized by decreased LV systolic function and increased LV mass, LV end-diastolic dimension (LVEDD), and LV end-systolic dimension (LVESD).(3,8,9) Similarly, a French study (N=22 BTHS patients) showed LV chamber size, LV mass, and LVEDD were increased, and LV ejection fraction (LVEF) was decreased during the first six months of life.(10)
In contrast, HCM is typically observed in adolescent/adult patients with BTHS and is characterized by thickened, stiff cardiac walls that prevent proper myocardial relaxation, resulting in reduced LV filling and stroke volume (LVSV),(2) a phenotype similar to heart failure with preserved ejection fraction (HFpEF). Consistent with the HCM phenotype and indicative of impaired LV diastolic function, negative LVSV z-scores were observed in patients 12 years of age and older in a subgroup analysis from a recently published cardiac natural history (NH) study in BTHS patients (Figure 1).(11)
Furthermore, a gradual decline in LVSV, and therefore in cardiac output, has been demonstrated in NH cohorts.(12) With findings suggesting an evolving cardiomyopathic phenotype, a recent longitudinal study in BTHS demonstrated: significant worsening of septal E:e’; LV global longitudinal strain and right ventricular fractional area of change; reduction in LV size/LVSV; and no change in LVEF over time.(13)
Echocardiography in BTHS consistently demonstrates that LV shortening fraction and LVEF are only mildly abnormal outside of infancy.(14) These findings support a temporal evolution of the cardiomyopathic phenotype in BTHS patients with advancing age from a DCM during infancy to one consistent with an HCM resembling HFpEF.
Therapeutic approach to cardiomyopathy in BTHS patients
The current approach to treating BTHS cardiomyopathy is primarily directed at alleviating symptoms, not targeting the underlying disease pathophysiology. Given the age-related-changing phenotype, a more logical therapeutic approach in BTHS adolescents/adults would be to treat as if the phenotype were that of HFpEF, focusing on improving LV relaxation and filling. The reduced LV volume and ensuing inadequate LV filling observed in HCM and HFpEF leads to decreased LVSV, an integral component of overall cardiac performance.(2) In the HFpEF phenotype, the LV must expand through improved LV relaxation to allow for adequate LV filling in order to increase LVSV.
Therefore, increased LV volumes and LVSV are appropriate goals for treating HCM of BTHS. In preclinical and clinical studies in heart failure (HF) and BTHS models, the mitochondria-targeting peptide, elamipretide, increases LVSV, LVEDV, and LVESV, making it a promising treatment option for HCM in BTHS.(2) Elamipretide readily penetrates and transiently localizes to the inner mitochondrial membrane (IMM), where it interacts with cardiolipin to improve IMM stability, enhance ATP synthesis, and reduce reactive oxygen species (ROS) production.(15-21) Cardiolipin is a logical therapeutic target because the primary defect underlying BTHS is altered cardiolipin content. Elamipretide is well-suited for BTHS because it interacts with the membrane bilayer, independent of the cardiolipin side chain composition, and interacts with accumulated monolysocardiolipin in a similar ratio.(21)
TAZPOWER phase 2 clinical trial
TAZPOWER evaluated the efficacy/safety of 12-week treatment with daily subcutaneous elamipretide in BTHS patients 12 years of age and older, followed by a 168-week open label extension (OLE) on elamipretide.(22) Similar to NH and longitudinal studies, the observed cardiac phenotype demonstrated poor LV active and passive filling, leading to reduced LVSV; small LVED and LVES volumes (both below the eighth percentile z-scores); low cardiac index (mean 2.3); and normal LVEF, all resembling the HFpEF type of LV failure.(2)
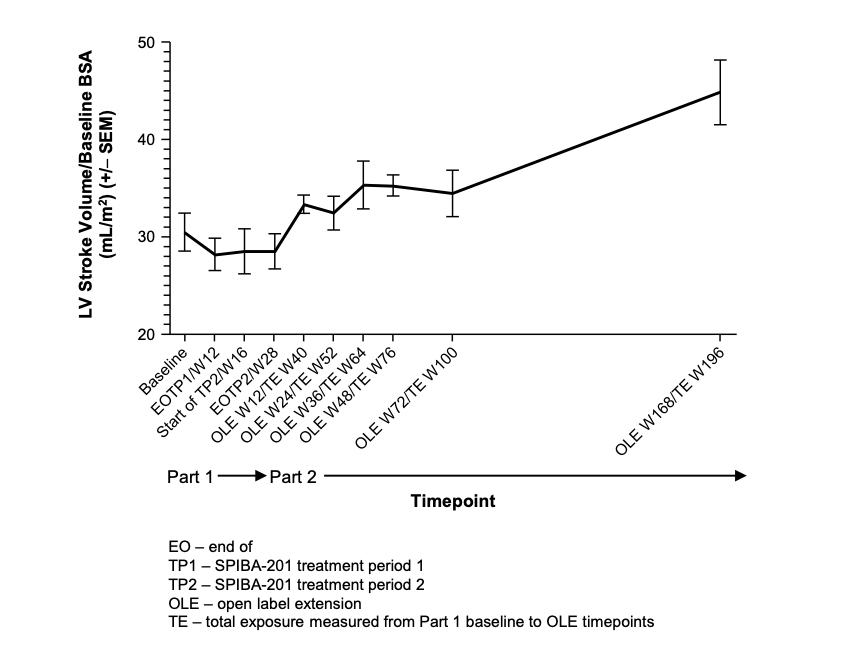
During the OLE portion, significant improvements in functional and cardiac outcomes were observed after elamipretide compared to baseline.(22) Elamipretide treatment resulted in a 16% improvement (30.5mL/m2 to 35.3mL/m2) and >45% improvement (mean 14.4mL; p=0.007) in LVSV indexed to body surface area (BSA) at 36 weeks and 168 weeks, respectively (Figure 2).(22) Significant improvements in LVED and LVES volumes indexed to baseline BSA were seen at all OLE time points.
Comparing TAZPOWER and OLE elamipretide-treated subjects, and a NH-study cohort of BTHS patients, it was demonstrated that the LVSV index increased significantly in elamipretide- treated patients versus NH controls.(12) Collectively, the TAZPOWER results support that long-term treatment with elamipretide may improve overall cardiac function, quality of life, and the long-term disease trajectory of BTHS.
Conclusions and future considerations
NH and longitudinal studies suggest the cardiomyopathic phenotype in BTHS transforms from DCM with reduced LVEF in infancy to HCM with preserved LVEF, similar to HFpEF, in adolescence and adulthood.(12,13,25) Treatment strategies and agents that improve LV relaxation, such as elamipretide, may be beneficial for use in adolescent and adult patients with BTHS considering the unique HCM phenotype. Understanding the temporal change in HF phenotypes in BTHS will enable the use of targeted treatments at the appropriate stages of the disease to improve cardiac performance and quality of life while preventing, or at the very least, retarding progression of the disease toward intractable end HF.
References
- Clarke SL, Bowron A, Gonzalez IL, et al. Barth Syndrome. Orphanet. J. Rare. Dis. 8, 23 (2013).
- Sabbah HN. Elamipretide for Barth syndrome cardiomyopathy: gradual rebuilding of a failed power grid. Heart Fail Rev. 27(5), 1911-1923 (2022).
- Spencer CT, Byrne BJ, Bryant RM, et al. Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am J Physiol Heart Circ Physiol. 301(5), H2122-2129 (2011).
- Wang G, Mccain ML, Yang L, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20(6), 616-623 (2014).
- Bertero E, Nickel A, Kohlhaas M, et al. Loss of mitochondrial Ca2+ uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in Barth Syndrome cardiomyopathy. Circulation. 144(21), 1694-1713 (2021).
- Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 356(11), 1140-1151 (2007).
- Cade WT, Bohnert KL, Peterson LR, et al. Blunted fat oxidation upon submaximal exercise is partially compensated by enhanced glucose metabolism in children, adolescents, and young adults with Barth syndrome. J Inherit Metab Dis. 42(3), 480-493 (2019).
- Spencer CT, Bryant RM, Day J, et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 118(2), e337- 346 (2006).
- Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 375(9716), 752-762 (2010).
- Rigaud C, Lebre AS, Touraine R, et al. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet. J. Rare. Dis. 8, 70 (2013).
- Sabbah H, Taylor C, Vernon H. Temporal Evolution of the Heart Failure Phenotype in Barth Syndrome and Treatment with Elamipretide. Future Cardiol. 19(4), 211-225 (2023).
- Hornby B, Thompson WR, Almuqbil M, et al. Natural history comparison study to assess the efficacy of elamipretide in patients with Barth syndrome. Orphanet. J. Rare. Dis. 17(1), 336 (2022).
- Chowdhury S, Jackson L, Byrne BJ, et al. Longitudinal observational study of cardiac outcome risk factor prediction in children, adolescents, and adults with Barth Syndrome. Pediatr. Cardiol. 43(6), 1251-1263 (2022).
- Roberts AE, Nixon C, Steward CG, et al. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med. Genet. A. 158A(11), 2726-2732 (2012).
- Sabbah HN, Gupta RC, Kohli S, Wang M, Hachem S, Zhang K. Chronic therapy with elamipretide (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ. Heart. Fail. 9(2), e002206 (2016).
- Sabbah HN, Gupta RC, Singh-Gupta V, Zhang K. Effects of elamipretide on skeletal muscle in dogs with experimentally induced heart failure. ESC. Heart. Fail. 6(2), 328-335 (2019).
- Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox. Signal. 11(9), 2095-2104 (2009).
- Szeto HH, Liu S, Soong Y, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 22(6), 1041-1052 (2011).
- Dai DF, Hsieh EJ, Chen T, et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart. Fail. 6(5), 1067-1076 (2013).
- Talbert EE, Smuder A J, Min K, Kwon OS, Szeto HH, Powers SK. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J. Appl. Physiol. 115(4), 529-538 (2013).
- Mitchell W, Ng EA, Tamucci JD, et al. The mitochondria- targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action. J. Biol. Chem. 295(21), 7452- 7469 (2020).
- Thompson WR, Hornby B, Manuel R, et al. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet. Med. 23(3), 471-478 (2021).
- Thompson WR, Manuel R, Abbruscato A, et al. Long-term Efficacy and Safety of Elamipretide in Patients with Barth Syndrome: 192-Week Open-label Extension Results of TAZPOWER. Presented at: American Society of Human Genetics (ASHG). October 25-29, 2022, Los Angeles, CA.
- Thompson WR, Manuel R, Abbruscato A, et al. Long-term Efficacy and Safety of Elamipretide in Patients with Barth Syndrome: Open-label Extension Results of TAZPOWER Through 192 Weeks. Presented at: Barth Syndrome Foundation (BSF). July 21-22, 2022, Virtual.
- Thompson WR, Manuel R, Abbruscato A, Carr J, Hornby B, Vernon HJ. Long-term Efficacy and Safety of Elamipretide in Patients with Barth Syndrome: 192-Week Open-label Extension Results of TAZPOWER. Presented at: United Mitochondrial Disease Foundation (UMDF) Annual Meeting. June 8-11, 2022, Phoenix, AZ.

This work is licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International.