The current primary treatment for hypophosphatasia is enzyme replacement therapy; however the development of a new gene therapy drug, ARU-2801, may change the landscape forever
Understanding hypophosphatasia
Hypophosphatasia (HPP) is a rare bone disease caused by mutations in the ALPL gene, which encodes tissue-nonspecific alkaline phosphatase (TNALP).
Patients can present with the following:
- Decreased serum ALP activity
- Hypocalcification of systemic bone
- Deformity of long bones
- Irregularity of the ends of the diaphysis
- Poor weight gain
- Seizures
Symptoms range from mild to severe depending on the patient and are classified into six types according to the age of onset: perinatal lethal form, perinatal benign form, infantile form, childhood form, adult form, and odontohypophosphatasia.
If untreated, almost all patients with the perinatal lethal form and about half of those with the infantile form will die.
The prognosis for other forms of the disease is good, but a variety of symptoms that can affect daily life can occur in all forms.
The current primary treatment for hypophosphatasia is enzyme replacement therapy. This method requires subcutaneous injections of enzymes 3-6 times a week, and the associated inflammatory reactions at the injection site can be problematic.
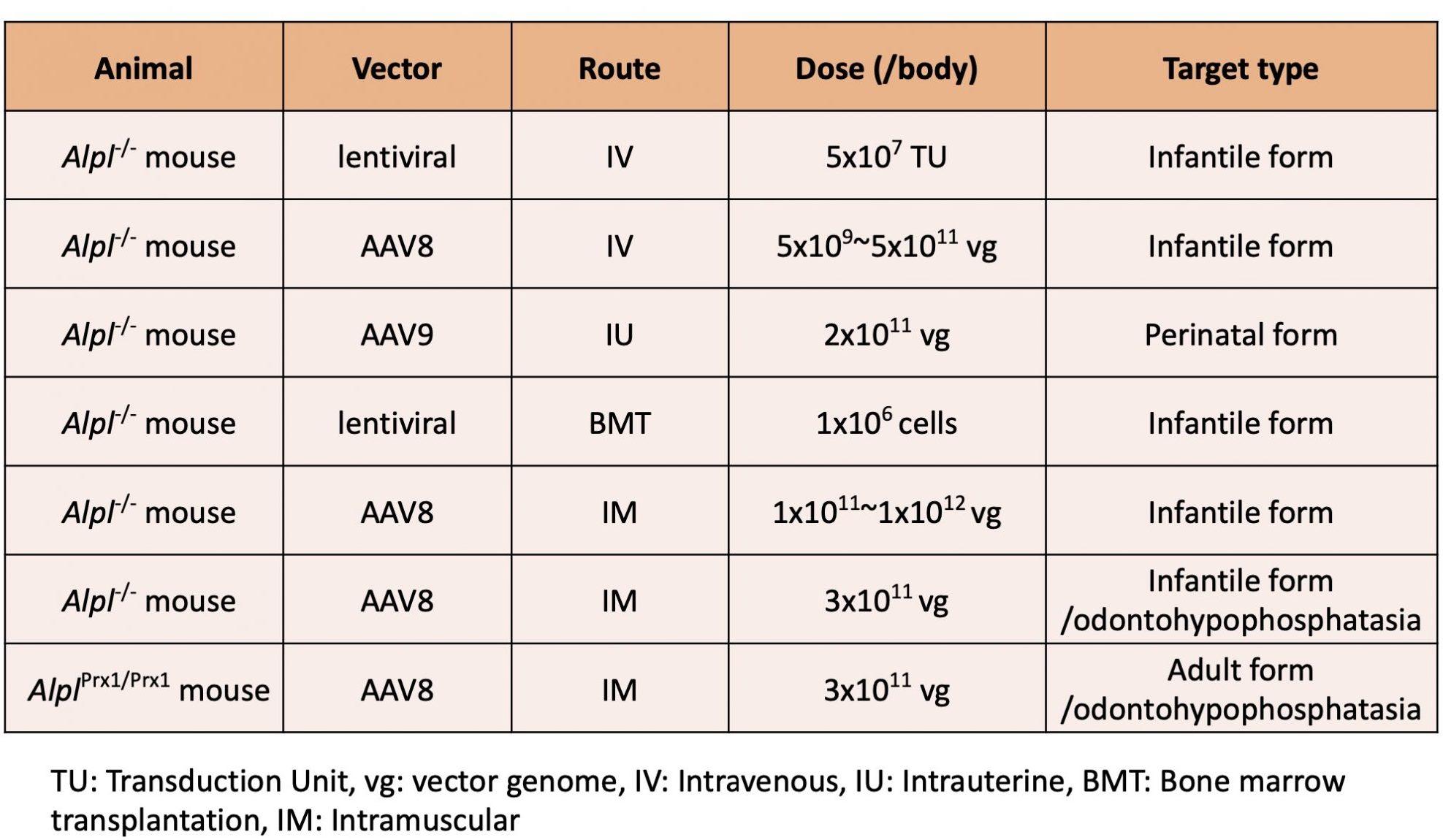
Gene therapy research for HPP
We have been studying gene therapy for HPP for many years and have tried a number of methods for treatment, such as:
- Intravenous administration of lentiviral vectors or adeno-associated virus (AAV) vectors expressing TNALP
- Intrauterine fetal administration of AAV vectors
- Transplantation of hematopoietic stem cells transduced with lentiviral vectors
- Intramuscular administration of AAV vectors
- Treating HPP infantile model mice lacking the Alpl gene (Alpl-/- mice), therapeutic effects such as prolongation of survival and improvement of bone formation have been observed.
Table 1 summarizes the gene therapy studies to date. Our results indicate that gene therapy is feasible for all types of hypophosphatasia, from perinatal to adult and odontohypophosphatasia.
Among these therapies, we are developing an intramuscularly administered TNALP-D10-expressing AAV type 8 vector (ARU-2801), which we believe to be the simplest and safest therapy for HPP.
Development of gene therapy drug (ARU-2801)
The ARU-2801 is a gene therapy drug that incorporates TNALP with bone affinity by binding D10 to the catalytic domain of human TNALP in an AAV vector.
The TTP (Target Product Profile) of ARU-2801 is shown in Table 2, and we believe that the development of ARU-2801 will provide long-term sustained efficacy with a single safe treatment and will be of great benefit to patients with HPP in terms of both ADL and quality of life.

Efficacy of ARU-2801
To evaluate the efficacy of ARU-2801 against HPP, we administered a single intramuscular dose of ARU-2801 to neonatal Alpl-/- mice and followed their plasma ALP activity, survival, and bone formation over an 18-month period. Plasma ALP activity was around 10 U/ml over a long period (18 months).
Untreated HPP mice died within 3 weeks, whereas ARU-2801-treated mice survived longer and showed weight gain, bone formation, and bone mineral density comparable to wild-type control mice.
In addition, intramuscular administration of ARU-2801 also improved alveolar bone, indicating that ARU-2801 is useful for the odontohypophosphatasia. Furthermore, in AlplPrx1/Prx1 mice, a late-onset (adult-type) HPP model, intramuscular administration of ARU-2801 also improved alveolar bone.
These results suggest that a single intramuscular administration of ARU-2801 provides a therapeutic effect in all forms of hypophosphatasia.
Examining the safety of ARU-2801
The safety of ARU-2801 was examined in Alpl-/- mice and non-human primates (NHPs) by administering a single intramuscular dose of ARU-2801.
Biodistribution of ARU-2801
Quantitative PCR analysis of the distribution of ARU-2801 in each organ showed that ARU-2801 was detected in the muscle of the administration side and in the skin of the administration site, but not in other organs.
Examination of tumor formation and carcinogenesis
No evidence of tumor formation or carcinogenesis was observed at necropsy, CT scan, or histopathology in both Alpl-/- mice and NHPs that were administered intramuscularly.
Immunotoxicity
No increase in ALT or AST, thrombocytopenia, anemia, or renal dysfunction was observed in Alpl-/- mice or NHPs treated with ARU-2801 intramuscularly during the observation period, and antibodies to TNALP-D10, a therapeutic protein, were produced after treatment with ARU-2801, and a temporary decrease in TNALP was observed, but no serious side effects were observed.
General toxicity
No abnormal calcification was observed in primates in gross findings, radiological examination, CT scan, and Von Kossa staining. No abnormal findings were observed in biochemical tests, including liver function, renal function, and Ca levels.
What does the future hold concerning the treatment for hypophosphatasia
As described above, ARU-2801 treatment for hypophosphatasia has been shown to be effective and safe in HPP mice and primates, and the single intramuscular administration of ARU-2801 avoids the adverse events reported in recent years with intravenous administration of AAV vectors.
ARU-2801 has been shown to be effective not only in prolonging the life of patients with hypophosphatasia but also in improving ADL, and quality of life. We are now preparing for the clinical trial of ARU-2801. We hope that ARU-2801 will be available to patients with hypophosphatasia as soon as possible.
- FOUNDING: This work was supported by JSPS KAKENHI Grant Numbers 15K09605 and 20K08268, and is supported by AMED under Grant Number JP23ek0109670.
- RESEARCH COLLABORATOR: Professor José Luis Millán, Human Genetics Programme, Sanford Children’s Research Centre
- COOPERATION COMPANY: Aruvant Sciences

This work is licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International.