Andrew Teschendorff, Professor at the Chinese Academy of Sciences, is developing computational systems-biological tools to identify cells at risk of turning cancerous
Predicting an individual’s risk of developing cancer is a huge challenge. With the exception of pediatric cancers and some adult cancer types that are caused by inherited genetic mutations (e.g., BRCA1 mutations in breast cancer), the great majority of cancer types are believed to be caused by genetic mutations that are acquired as we age, following prolonged exposure to cancer risk factors like smoking, inflammation or viral infections. These genetic mutations alter the underlying DNA sequence, altering gene and protein function and hence also cell function. We now know, however, that other types of molecular alterations, referred to as ‘epigenetic changes,’ also contribute to cancer development. One of these is DNA methylation, where the chemical bond of particular sites in the DNA is modified by the attachment of a methyl group (a carbon atom with three hydrogen atoms). DNA methylation does not alter the underlying DNA sequence, but it can nevertheless affect the activity of a nearby gene and, hence, alter a cell’s normal function. Thus, predicting an individual’s risk of cancer would require us to measure all genetic and epigenetic changes in every tissue of the human body, which is currently impossible. However, some organs are accessible, and clinical samples representative of the tissue can be obtained through routine screening programs: for example, colonic polyps are routinely removed via colonoscopy since polyps are a common precursor lesion of colon cancer. Collecting such clinical specimens from precancerous lesions and subsequently examining the cells from these lesions may thus make it possible to quantify a person’s cancer risk.
CancerStemID
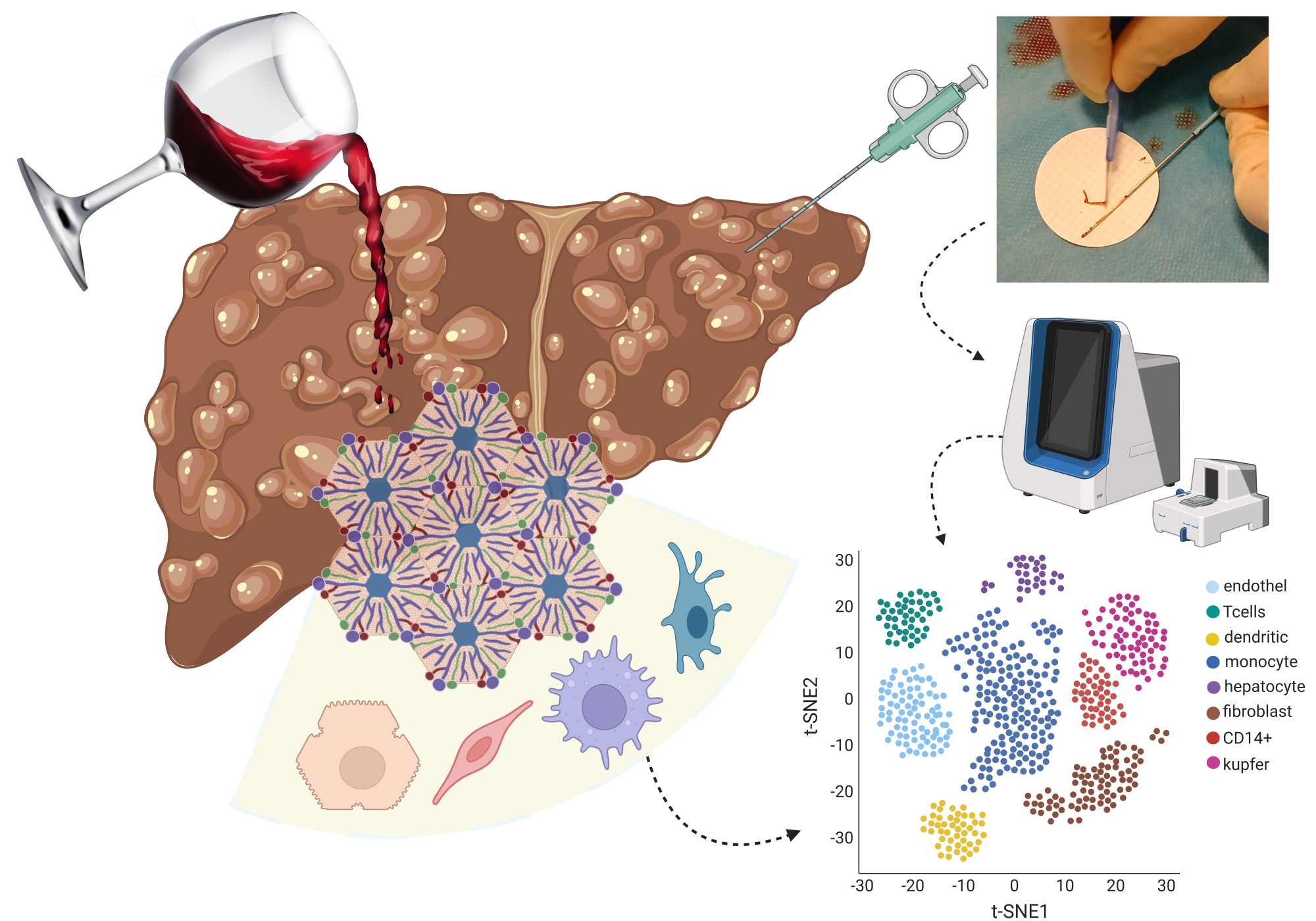
Advances in biotechnology now allow measurement of the activity (termed ‘mRNA expression’) of all human genes in single cells from precancerous lesions, but how to quantify a cell’s cancer risk from such gene mRNA expression patterns is non-trivial. Andrew Teschendorff, working at the Shanghai Institute for Nutrition and Health, which is part of the Chinese Academy of Sciences, has recently developed a computational tool called CancerStemID, (1-3) which allows cancer risk to be quantified at the resolution of single cells.
The key idea underlying CancerStemID is simple yet powerful. Andrew Teschendorff hypothesized that there are key genes called tissue-specific transcription factors that, if disrupted genetically or epigenetically and in sufficient numbers, could rewire a cell’s normal function, endowing this cell with an increased and aberrant phenotypic plasticity and entropy, (4,5) which would, in turn, increase the risk of this cell falling into a cancer state. Normally, tissue-specific transcription factors are genes that need to be switched on in a given tissue or organ for that organ to function properly, a cellular state usually referred to as ‘differentiated.’ In precancerous lesions, a number of these tissue-specific transcription factors would be switched off due to a genetic or epigenetic alteration that, in effect, ‘inactivates’ these transcription factors. CancerStemID is a mathematical algorithm that allows one to estimate the activity of these tissue-specific transcription factors in each individual cell of a precancerous lesion. In collaboration with Professor Chen Wu from the Chinese Academy of Medical Sciences, Andrew Teschendorff showed that the number of inactivated tissue-specific transcription factors increases with cancer progression and that this number could serve as a cancer-risk score. Cells with a particularly high number of inactivated tissue-specific transcription factors resemble stem cells but are also distinct from them in that the inactivation of the transcription factors is irreversible, whilst, in normal ‘healthy’ stem cells, these same transcription factors are suppressed by a different epigenetic mechanism that is dynamic and reversible. That CancerStemID could identify the cells at higher cancer risk was demonstrated in the context of non-cancerous lesions that precede esophageal squamous cell carcinoma, a very aggressive form of cancer with a dismal outcome. However, the concept underlying CancerStemID appears to work for every cancer type tested so far, including colon, stomach, and lung. (6,7)
The CancerStemID study is also significant in demonstrating what causes the inactivation of these tissue-specific transcription factors. It has long been thought that cancer is mainly driven by genetic mutations, but according to Teschendorff, if we focus only on tissue-specific transcription factors, the main alteration affecting these specific genes is DNA methylation, which accumulates with age in the promoter regions of these transcription factors, causing their inactivation.
To conclude, we are at an exciting juncture in the cancer risk prediction field, with the growing importance of epigenomics in cancer development now widely accepted and with advances in single-cell data biotechnologies offering unprecedented opportunities to not only deepen our system’s biological understanding of cancer genesis but to also develop novel strategies for personalized prediction of cancer risk.
References
- Liu, T. et al. Computational Identification of Preneoplastic Cells Displaying High Stemness and Risk of Cancer Progression. Cancer Res 82, 2520-2537 (2022).
- Teschendorff, A.E. Computational single-cell methods for predicting cancer risk. Biochem Soc Trans 52, 1503-1514 (2024).
- Teschendorff, A.E. CancerStemID: one step closer to predicting cancer risk? (Research Pod, https://www.youtube.com/watch?v=r42e-vI6m7s, 2024).
- Teschendorff, A.E. & Enver, T. Single-cell entropy for accurate estimation of differentiation potency from a cell’s transcriptome. Nat Commun 8, 15599 (2017).
- Teschendorff, A.E. & Feinberg, A.P. Statistical mechanics meets single-cell biology. Nat Rev Genet 22, 459-476 (2021).
- Maity, A.K. & Teschendorff, A.E. Cell-attribute aware community detection improves differential abundance testing from single-cell RNA-Seq data. Nat Commun 14, 3244 (2023).
- Teschendorff, A.E. & Wang, N. Improved detection of tumor suppressor events in single-cell RNA-Seq data. NPJ Genom Med 5, 43 (2020).