With a particular focus on glioma, Dr Peter J Houghton from Greehey Children’s Cancer Research Institute outlines the barriers that have hindered the development of effective therapies for childhood solid cancers
Childhood cancer represents less than 1% of human cancers, thus, is not a priority for drug development in the pharmaceutical industry. However, the consequences of having a child diagnosed with a malignant disease can be devastating for families. Developing new therapies for childhood solid cancers presents certain constraints that are seldom encountered with neoplastic diseases diagnosed in adults.
Childhood tumours are rare, which has therefore restricted large-scale drug evaluations or randomised clinical trials. For example, of the new Phase I agents evaluated in adult malignancies, less than 30% receive an adequate assessment in children.
Furthermore, drug-screening strategies focus on selecting new anti-cancer agents with specific activity against adult neoplastic diseases (e.g. colon, lung, breast, etc.) so that agents with specific activity against childhood malignancies might not be identified.
A further restriction on drug development is that many ‘common’ solid cancers and tumours seen in children respond to drugs of established efficacy, resulting in many patients being cured. This ethically precludes the use of ‘experimental’ agents in many untreated cases.
Poor prognosis of advanced or metastatic solid cancers
Advanced or metastatic solid cancers in children have a poor prognosis. For most patients with metastatic disease at diagnosis, the long-term event-free survival (EFS) is less than 30%.
Current therapies include all the known active agents plus radiation, but neither the intensification of chemotherapy by increasing the dose of drugs or adding active agents to standard agents nor the use of high-dose chemotherapy with stem-cell rescue have improved outcomes over the last 30 years.
Certainly, these data question the value of continuing to develop classical cytotoxic therapies. However, genomic studies have revealed few mutations that can be exploited therapeutically.
Challenges in developing effective therapies for solid cancers
One of the major problems in developing effective therapies that do not lead to resistance is that we have very little knowledge of changes in tumour characteristics during the evolution of resistance under therapeutic stress.
Rarely are tumours biopsied at relapse; hence mutations conferring resistance are not documented, and the characteristics of the relapse tumour are largely unknown. Genetic changes have been shown to confer drug resistance to cytotoxic agents through gene amplification or point mutations in genes encoding the drug target.
However, much less is understood about resistance mechanism(s) selected for very complex multi-drug regimens. There are a multitude of reasons why drug therapy fails in advanced diseases. Drugs can be excluded from brain tissue by the blood-brain barrier (BBB) for brain tumours, exacerbating drug failure.
Brain tumours constitute about 21% of paediatric cancers; low-grade tumours occur most frequently. While the survival rate for these patients is relatively good, even at five years, these indolent tumours progress.
Current treatments and treatment-resistant progressive disease
Current treatment of glioma includes intensive chemo-radiation therapy that leads to cognitive decline, malignant transformation, and other life- debilitating or threatening sequelae.
The 15-year incidence of adverse outcomes such as blindness, hearing loss, obesity, and hormonal imbalance was 18%, 22%, 53% and approximately 25%, respectively. Among survivors assessed for intellectual function, 34% had an intelligence quotient (IQ) below average.
Treatment-resistant progressive disease is the most common cause of death.
Genomic studies have shown that in low-grade tumours (juvenile pilocytic astrocytoma), ~90% have a tandem duplication involving the KIAA1549 and BRAF genes that generate constitutively active KIAA1549-BRAF fusion. Higher-grade tumours tend to have activating point mutations of BRAF, most frequently the V600E variant (where glutamic acid is substituted for the naturally occurring valine).
Such activating mutations are identified in diffuse astrocytomas (23%), gangliogliomas (33%), and pleomorphic xanthoastrocytoma (70%). Some 10-15% of paediatric glioblastomas and many epithelioid glioblastomas also carry this mutation. It is estimated that around 1400 new paediatric BRAF mutant brain tumours are diagnosed annually in the US.
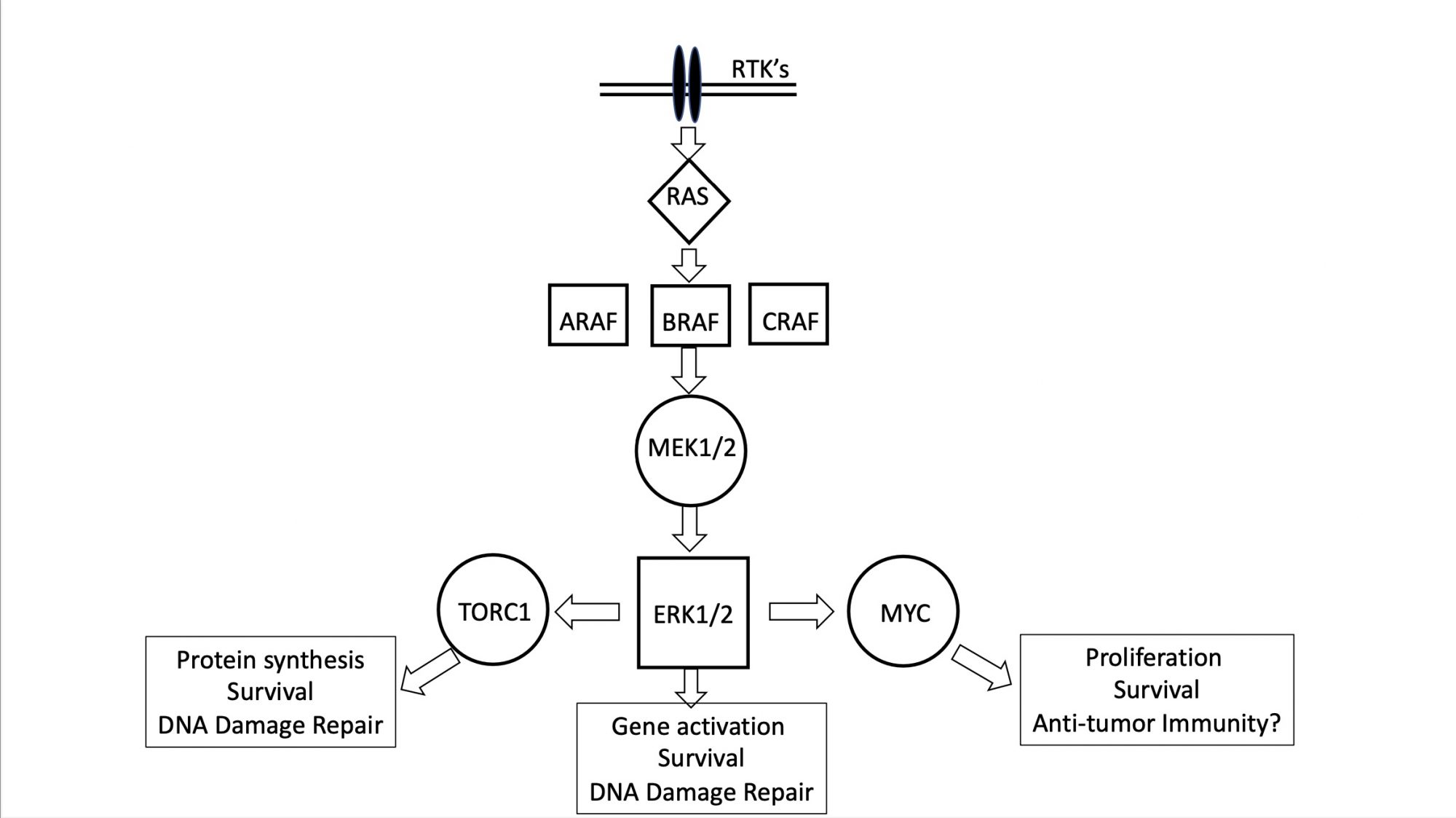
The MAPK pathway
A common characteristic of these solid cancers and tumours is receptor tyrosine kinase (RTK) activation of the MAPK pathway, highly simplified in the Figure. Low-grade glioma activation of BRAF abrogates the requirement for growth factor-induced signalling activation.
First-generation BRAF inhibitors (BRAFi) are not active against tumour cells with the KIAA1549-BRAF fusion, and preclinical data from childhood astrocytoma xenografts in immune-deficient mice were important in demonstrating selective anti-tumour activity of selumetinib (a MEK inhibitor that blocks signalling downstream of mutant BRAF) only in BRAF(V600E) mutant astrocytoma.
Such data was used to develop Phase I (PBTC029) and subsequent selumetinib Phase II trials of selumetinib that demonstrated high-level activity, with around 40% of patients having tumour control over two years.
Could selumetinib be an alternative?
These clinical results show that selumetinib could be an alternative to standard chemotherapy and led to two COG Phase 3 studies comparing standard chemotherapy to selumetinib in patients with newly diagnosed LGGs. In the Phase 2 trial, treatment was limited to two years as the long-term consequences of MEK inhibitors in children were unknown.
In those patients with tumours driven by KIAA1549-BRAF fusions, a proportion did not progress when treatment was terminated, or the drug dose decreased after two years, suggesting possible cures. However, most BRAF(V600E) mutant tumours either progress on treatment (i.e., become resistant during therapy) or rapidly when selumetinib is terminated.
These tumours remain a clinical challenge, as do higher-grade gliomas that are a significant focus for research and drug development. The focus on developing more effective therapies for these high-risk BRAF-mutant gliomas will depend on understanding why tumours become resistant to MAPK inhibitors and developing strategies that reverse or, perhaps, more importantly, prevent the development of resistance.

This work is licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International.