
In 1980, hundreds of individuals reported to emergency clinics in Northern California complaining of tremors, motility problems, and mild cognitive deficits – all classic signs of Parkinson’s disease (PD)
PD is a slowly progressing neurodegenerative disease associated with aging; however, these patients were mostly young and had developed symptoms virtually overnight.
It was eventually discovered that they shared a common clinical history: exposure to an illicit synthetic heroin contaminated with the chemical 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, or MPTP. Subsequent studies in experimental animals confirmed that MPTP could cause sudden “parkinsonism”, including not only motor behaviors, but also changes in the brain that resembled that of PD.
The incident with MPTP was a landmark event in our understanding of the causes of PD because it alerted scientists to the potential role of environmental chemicals in promoting PD.
Up until then, it was thought that PD was predominantly a genetic disease. While several familial forms of PD are known to be associated with specific gene variants, genetic alterations are estimated to account for only 5–10% of PD cases.
It is now the general consensus in the field that most cases of PD are triggered by a complex interplay of genetic and environmental risk factors. Here, we discuss the evidence linking chemicals in the human environment to PD risk.
What is PD and why do we think environmental chemicals and PD are linked?
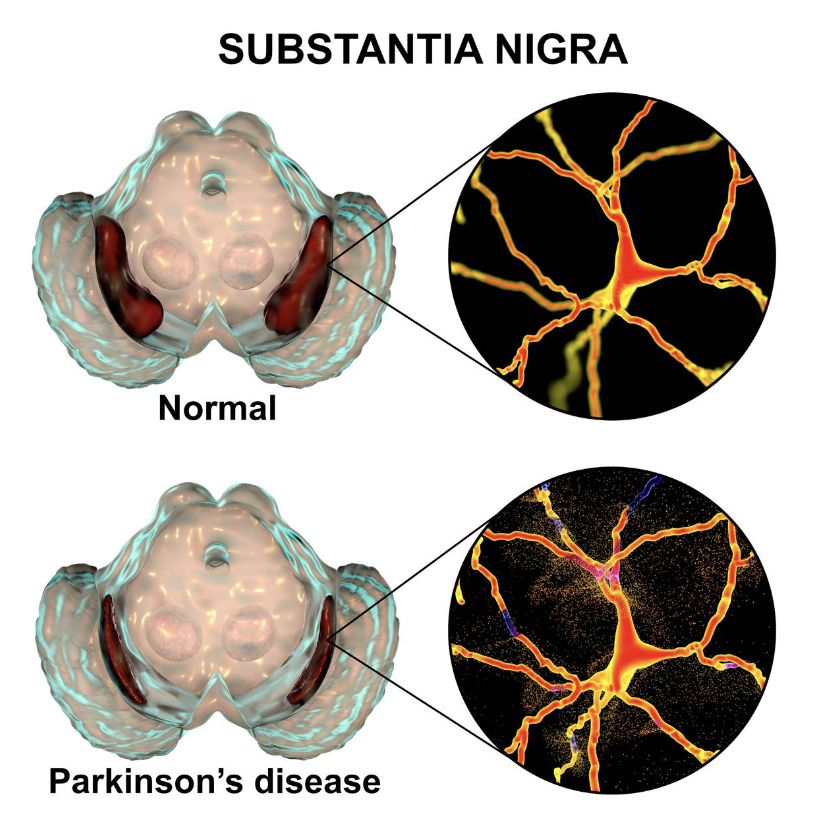
PD is a brain disorder that causes gradual loss of muscle control. Distinctive symptoms of the disease include tremors, stiffness, slowed body movements or bradykinesia, and poor balance. PD is caused by neurodegeneration (the atrophy or loss of neurons) in a small area of the brain called the substantia nigra (Figure 1).
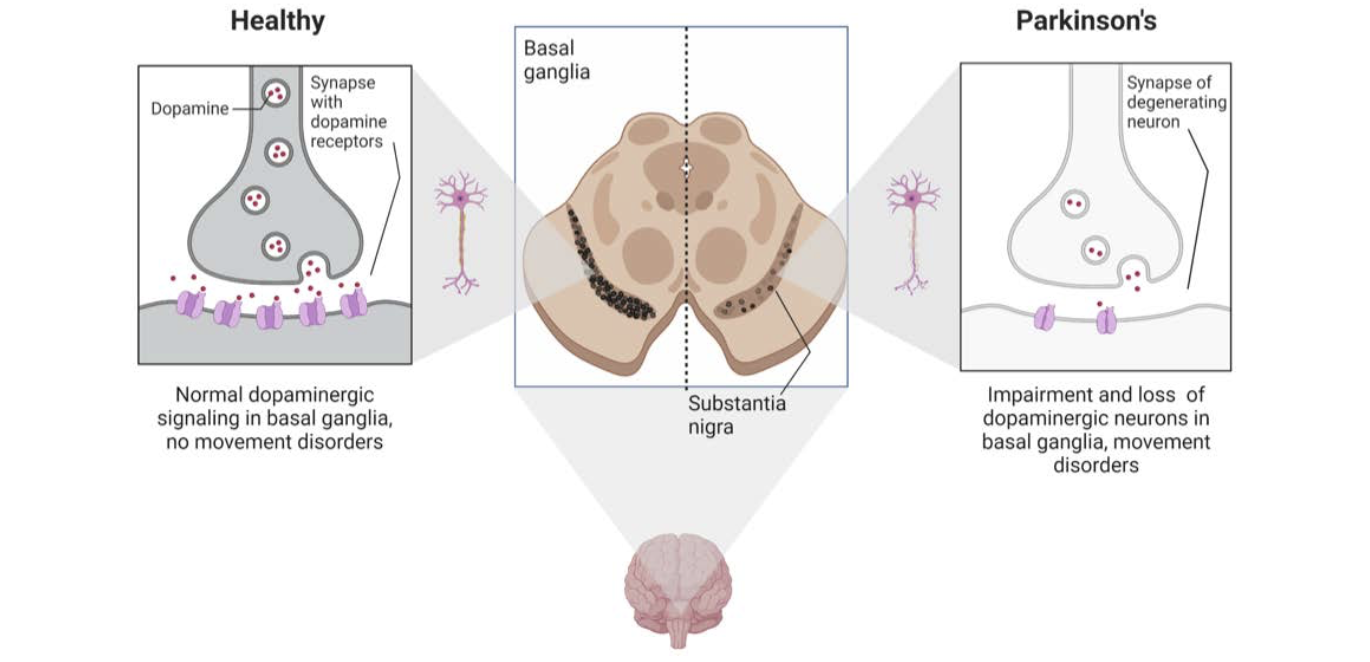
The substantia nigra controls motor function via release of the neurotransmitter dopamine. The progressive loss of dopamine-producing, or dopaminergic, neurons results in progressive dysfunction of the neural circuits that control motor behavior. PD often includes other non- motor effects, including deficits in memory and attention, gastrointestinal problems, depression, sleep disorders, and anxiety.
When Dr. Parkinson first described PD in 1817, it was identified as the cause of death of approximately 22 people out of a population of 15 million in England and Wales. Due to increased awareness of the disease and improved diagnostic tools, it now appreciated that PD affects a significant number of individuals and that these numbers are increasing. Worldwide, up to 10 million people suffer from PD, and approximately 60,000 more Americans are diagnosed with PD every year. According to U.S.
Census Bureau projections, 680,000 people in the U.S. over the age of 45 had PD in 2010, whereas in 2020, the number had risen to approximately 930,000, and by 2030, the number is expected to be 1,238,000. Currently available treatments can slow the progression of PD symptoms, but there are no treatments that either prevent or cure PD.
These sobering statistics underscore the need to identify factors that influence individual risk for PD. Environmental risk factors are of particular interest because unlike genetic factors, which currently cannot be readily corrected, environmental risk factors are modifiable. Identification of specific environmental factors that increase or decrease PD risk would inform strategies for decreasing PD risk by decreasing exposure at not only the individual level, but also the population level.
Public health efforts to mitigate risk at the population level are likely to have the broadest impact, because they can affect even those individuals who do not have the means to access individualized medical treatments or change their lifestyles.
Numerous epidemiological studies during the past two decades have identified positive associations between PD and several environmental factors, including exposure to neurotoxic pesticides/herbicides and heavy metals (Table 1) as well as traumatic brain injury. Other environmental factors that have been implicated as potential risk factors for PD include industrial chemicals, wood pulp mills, farming, well-water consumption, and rural residence.
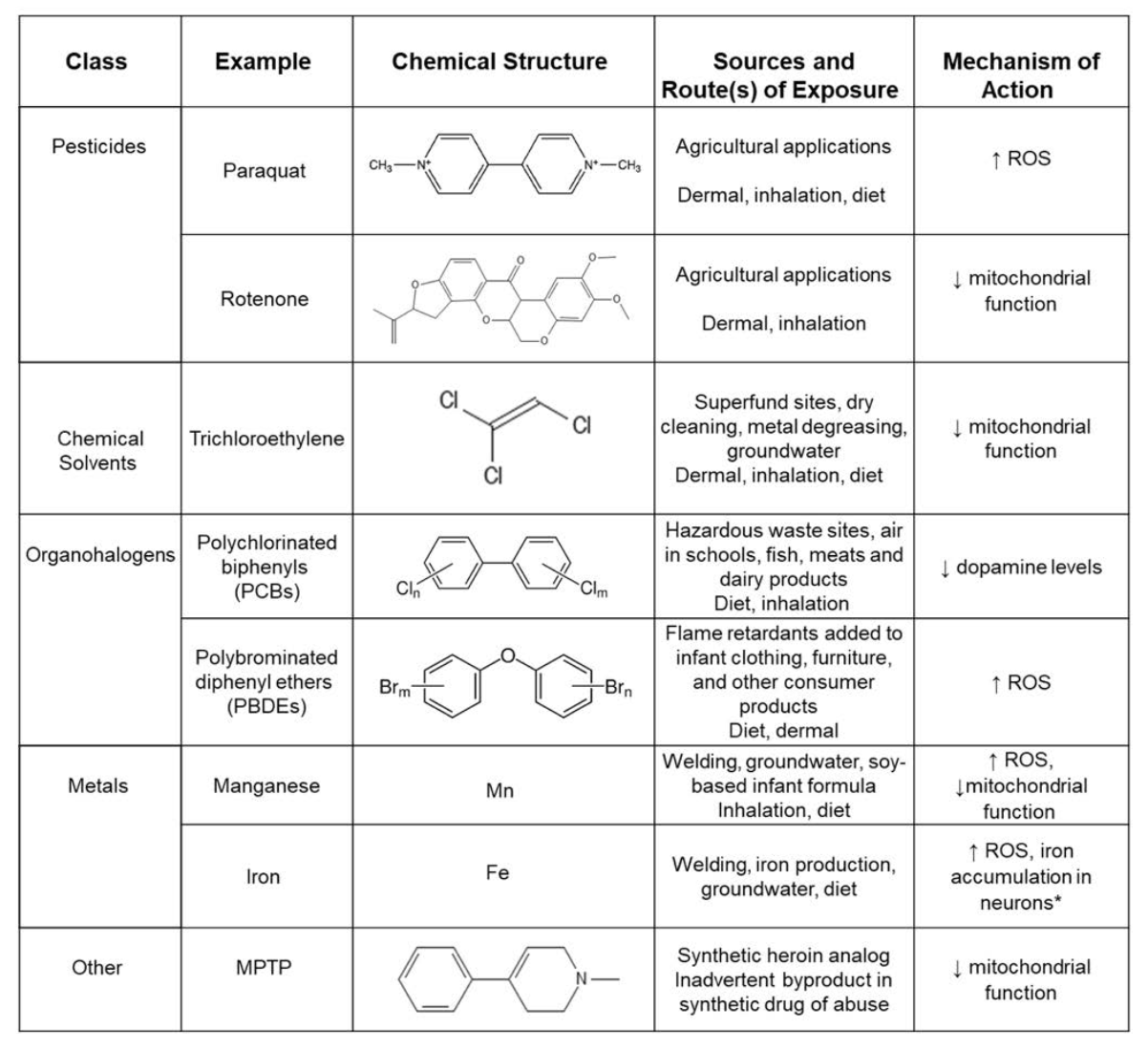
Environmental chemicals associated with increased PD risk
Manganese:
Manganese “Mn” is an essential metal needed to support diverse biological processes, including bone growth, reproduction, metabolism, cellular antioxidant response, and the function of many enzymes. The levels of manganese in the typical human diet are below the threshold for toxicity, but toxic manganese exposures can result from ingestion of contaminated well water, which is not uncommon in various geographical locations across the world, or soy-based infant formulas.
Exposure to toxic levels of Mn can also occur via inhalation of volatilized Mn fumes in occupatonal settings, such as in welding, mining, steelwork, and battery manufacturing.
Only 20 years after Dr. Parkinson’s initial characterization of PD, the first symptoms of Mn toxicity were documented in an essay by John Couper. He described cognitive dysfunction, disturbed gait, muscle weakness and facial paralysis in factory workers exposed to Mn primarily via inhalation.
Documentation of tremors and impaired motor function amongst workers occupationally exposed to Mn continued into the early 20th century. It is now well established that chronic exposure to high levels of Mn can cause manganism, a neurodegenerative disorder similar to PD characterized by bradykinesia, muscle rigidity, unstable posture, and an impaired gait. Loss of dopaminergic neurons in the substantia nigra has been reported in workers with manganism, although Mn also impacts dopaminergic neurons in other brain regions, and neuronal cell types other than dopaminergic neurons. The symptoms of manganism also do not respond to treatments used to slow the progression of PD.
There is also evidence that Mn is associated with an increased risk of true PD as well. A higher PD incidence was found in urban counties with high levels of Mn in the drinking water, and several studies have connected both welding and chronic consumption of Mn-contaminated drinking water to increased PD risk.
Paraquat:
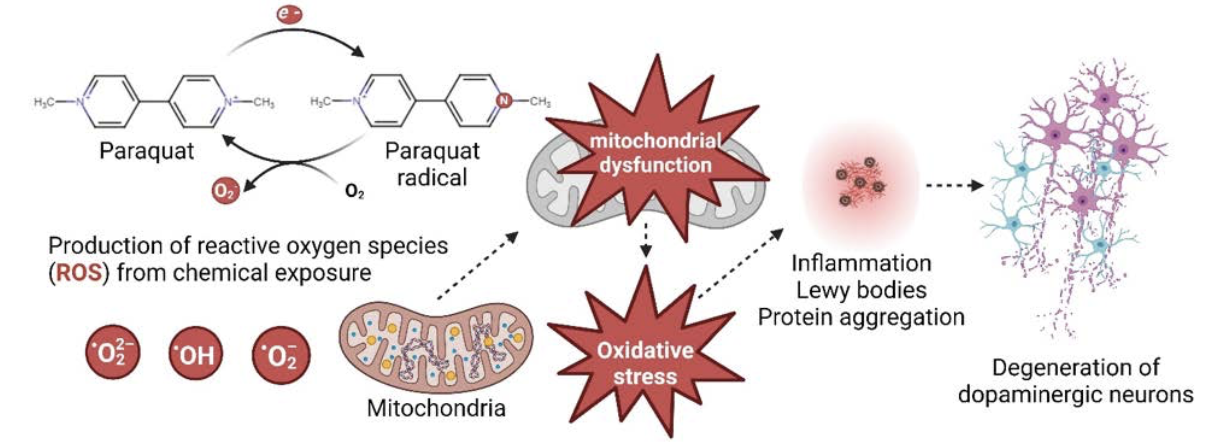
Paraquat is a neurotoxic herbicide that is still widely used around the world to control annual and perennial weeds. Its structure is similar to that of MPTP (see Table 1) and in experimental animal models, paraquat has been shown to induce alpha-synuclein aggregation and trigger neurodegeneration of dopaminergic neurons.
Interestingly, dopaminergic neurons appear to be uniquely sensitive to the neurotoxic actions of paraquat because the herbicide is taken up into neurons via the dopamine transporter, a transmembrane protein that normally functions to transport dopamine back into dopaminergic neurons after it has been released into the synapse. Paraquat is a highly oxidizing molecule that readily generates oxygen free radicals, which in turn cause oxidative stress, that can promote neuronal cell death.
It has long been thought that humans are typically exposed to paraquat via inhalation and dermal absorption. In cases where proper personal protective equipment (PPE) is worn to protect against dermal exposure, inhalation of paraquat rarely results in levels that exceed the safety threshold limit established by the National Institute for Occupational Safety and Health (NIOSH).
Thus, dermal exposure appears to be the more critical route of toxic exposure, and dermal exposure to high levels of paraquat may cause potentially fatal systemic poisoning. Emerging evidence suggests that ingestion may also be an important route of exposure. Women who did not live or work on a farm but who drank community well water in areas of heavy paraquat use, were reported to have significantly higher tissue levels of paraquat than control subjects, and up to 55% of their newborns also had measurable paraquat concentrations.
While there are epidemiological data indicating that paraquat exposure is associated with
a two-fold increased risk of developing PD, the link between paraquat and PD remains a controversial subject. Discrepant findings across epidemiological studies may be due in part to the reliance of self-reporting to estimate paraquat exposure in a number of these studies.
Self-reporting relies on recall memory of specific pesticide use, which may not always be accurate.
It has also been suggested that the inconsistent epidemiological data support a model in which the influence of paraquat on PD risk depends upon co-exposure with one or more other common environmental chemicals linked to increased PD, such as the pesticide, Maneb. In addition, there is evidence that genetic factors modify the effect of paraquat on PD risk.
For example, variations in the GSTT1 gene the enzyme glutathione S-transferase theta 1, which is a critical component of the cell’s antioxidant system, were found to influence PD risk associated with paraquat exposure. Individuals with a GSTT1*0 genotype had a 7.4-fold greater risk of developing PD than those with a GSTT1*1 genotype when exposed to paraquat. Metabolic genetic variants also appear to significantly change an individual’s risk of PD associated with paraquat exposure.
Given the concerns regarding paraquat’s neurotoxic potential, a number of countries have issued regulations to limit paraquat exposures. For example, in the U.S., the Environmental Protection Agency (EPA) issued an interim decision in 2020 that proposed more stringent protections to reduce human exposure, while paraquat use has been banned in several European countries, including the UK, since 2007.
Trichloroethylene:
PD has been linked to various chemical solvents, with perhaps the strongest association established for the solvent, trichloroethylene (TCE). First synthesized in 1864, TCE was used extensively worldwide through the 1900s for diverse applications ranging from the extraction of vegetable oils to the decaffeination of coffee to anesthetic use. Up until the 2000’s TCE was also used for dry cleaning and as a spot cleaner. However, the majority (up to 85%) of TCE produced in the U.S. and many European countries was used for metal degreasing.
Because of TCE’s widespread use, human exposure was common until the early 2000s. Today, exposures are predominantly occupational, although potentially toxic levels of TCE have been detected in soil, groundwater and indoor air at Superfund sites, hazardous waste dump sites and waste burn sites. For example, TCE was the primary contaminant found in the drinking water of Camp Lejeune, a military training facility located in North Carolina in the U.S. Numerous Camp Lejeune employees were exposed to TCE released by the Hadnot Point water treatment plant from 1950 to 1985.
This water system, which supplied drinking water to the facility, had become contaminated with TCE due to chemical dumping, leaking storage tanks, and improper waste disposal. TCE levels detected in the water were 1,400 μg/L, which is 280 times greater than the current U.S. maximum allowable contaminant level of 5 μg/L. A retrospective health outcome study of Camp Lejeune employees employed between 1973 and 1985 was conducted, with follow-ups starting in 1979 and ending in 2008. This group was compared to a similar military facility, Camp Pendleton, whose employees were not exposed to TCE-contaminated water.
Camp Lejeune employees had over twice the chance of developing PD than Camp Pendleton employees and were at an increased risk of developing certain cancers, such as kidney, liver and non-Hodgkin’s lymphoma.
Researchers also found a significant loss of dopaminergic neurons in the brains of Camp Lejeune employees chronically exposed to TCE.
Epidemiological studies of other cohorts have similarly found an association between PD and TCE. A longitudinal study of male twin pairs born between 1917 and 1927 who served in the military and in which at least one twin exhibited signs of PD indicated a link between TCE exposure and increased risk of developing PD.
In a study of industrial workers, three workers that directly handled TCE developed PD while those who worked farther from the source of TCE, but still inhaled the chemical, exhibited some limited PD-related pathologies. Based on these studies, and animal studies confirming that exposure to human-relevant levels of TCE caused loss of dopaminergic neurons and motor dysfunction, the EPA proposed to ban TCE for commercial and consumer use in December 2016.
Outstanding data gaps
There is now compelling evidence that chemicals in the human environment can increase individual risk for PD. However, there remain a number of critical, unanswered questions. An obvious data gap is an understanding of which of the 80,000+ chemicals to which humans are exposed confer risk for PD. As a first step toward addressing this data gap, it will be necessary to develop high-throughput assays that leverage biologically relevant cell culture models and/or simple animal model systems, such the nematode, Caenorhabditis elegans, or the zebrafish, Danio rerio, to screen large numbers of chemicals for their potential to cause PD-relevant phenotypes.
Another critical question is how do diverse chemicals promote the development and/or progression of PD? Insight on this question was initially provided by studies of the mechanism(s) underlying MPTP-induced parkinsonism. MPTP is a small, uncharged molecule that can readily cross the blood-brain barrier to move from the systemic circulation into the brain. Once in the brain, MPTP, which itself is not neurotoxic, is metabolized to MPP+ by the enzyme monoamine oxidase-B (MAO-B) in glial cells. MPP+, a charged molecule that cannot cross the blood-brain barrier back into the systemic circulation, bears significant structural similarity to dopamine.
Thus, MPP+ is rapidly taken up into dopaminergic neurons by the dopamine transporter. Once in neurons, MPP+ diffuses into the mitochondria where it directly inhibits mitochondrial complex I to decrease ATP production and increase release of reactive oxygen species (ROS).
Dopaminergic neurons are under intense bioenergetic demand, which makes them more vulnerable to mitochondria deficits and oxidative stress compared to non-dopaminergic neuronal cell populations. This may explain why these environmental chemicals selectively target the neuronal cell type implicated in PD.
Oxidative stress caused by environmental chemicals increases intracellular calcium levels, and studies using cell culture models have demonstrated that transient increases of intracellular calcium may induce formation of intracellular aggregates of α-synuclein. If a similar process occurs in the intact brain, it might explain how PD-associated chemicals promote the formation of Lewy bodies, the characteristic pathological lesion observed in PD.
Other chemicals and their relation to PD
Other chemicals that have been linked to increased PD risk are also known to increase ROS and/or decrease mitochondrial function (Table 1). For example, the pesticide rotenone directly inhibits the mitochondrial respiratory chain, resulting in energy deficits and oxidative stress (Table 1).
Other PD-associated environmental chemicals, such as paraquat (Figure 2), Mn and iron (Fe) induce oxidative stress and impair mitochondrial function via other mechanisms. Alternatively, polychlorinated biphenyls (PCBs) are thought to promote PD pathogenesis by decreasing dopamine levels in the brain (Table 1). There is also emerging evidence that environmental chemicals may influence PD risk independent of direct effects on the brain, acting instead on peripheral targets, such as the gut microbiome.
We have a limited understanding of how environmental chemicals interact with
genetic PD risk factors to influence individual risk of developing PD. One prevailing hypothesis is that both environmental chemicals and genetic factors interfere with the same process or signaling pathway in the brain to amplify each other’s impact.
The combined effect then pushes the system above the threshold for disease initiation. Alternatively, it has been proposed that genetic factors may modify metabolic pathways that either toxify or detoxify the chemical risk factor, thereby effectively changing the level of the toxic chemical in the brain. While there is experimental evidence to support both of these possibilities, what we know is significantly dwarfed by what we don’t know, including the identification of specific gene-environment interactions of relevance to PD.
Conclusion
Dr. Parkinson’s initial characterization of PD coincided with the advent of the industrial and Chemical Revolution. In the decades since, industrialization has increased globally, leading to the development and production of diverse chemicals in large quantities. Pesticides, metals, and chemical solvents are among the classes of chemicals that have been identified as potential PD risk factors, but undoubtedly there are many more. Absent a cure for PD, perhaps the best course of action is to develop public health interventions that reduce exposures to environmental chemicals associated with PD.
There is evidence of the potential impact of such an approach. For example, the introduction of smoke-free legislation contributed to a 10% decrease in childhood asthma. Other efforts to reduce air pollution have been attributed to a decrease in respiratory and cardiovascular mortalities and morbidities. Therefore, if we can identify the chemicals that modify PD risk, and determine which genetic factors increase susceptibility to specific PD-associated chemicals, we stand a chance of significantly decreasing the number of individuals who develop this disease.
Additional Resources for Further Reading
- Huang M, Bargues-Carot A, Riaz Z, Wickham H, Zenitsky G, Jin H, Anatharam V, Kanthasamy A, Kanthasamy AG (2022) Impact of environmental risk factors on mitochondrial dysfunction, neuroinflammation, protein misfolding, and oxidative stress in the etiopathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 23: 10808. https://doi. org/10.3390/ijms231810808
- Dorsey ER, Zafar M, Lettenberger SE, Pawlik ME, Kinel D, Frissen M, Schneider RB, Kieburtz K, Tanner CM, De Miranda BR, Goldman SM, Bloem BR (2023) Trichloroethylene: An invisible cause of Parkinson’s disease? Journal of Parkinson’s Disease. 13(2): 203-218. doi: 10.3233/JPD-22504
- McKnight S, Hack N (2020) Toxin-induced Parkinsonism. Neurologic Clinics. 38(4): 853-865. https://doi.org/10.1016/j.ncl.2020.08.003