
Michael Roth, Research Group Leader, Pulmonary Cell Research at University Hospital Basel & University Basel sheds light on inflammatory lung diseases
The prevalence of asthma, chronic obstructive pulmonary disease (COPD), and other lung diseases are increasing worldwide and represent a major issue for daily life for all patients. According to the World Health Organization, 280 million people suffer from asthma and up to 40% of children suffer from asthma symptoms.
In 2015, 65 million people were diagnosed with severe COPD with a death rate of 3 million per year. Both diseases are characterised by chronic airway inflammation, which can be controlled by pharmaceutical drugs and airway wall remodelling, which is insensitive to drugs. The American Thoracic Society stated in 2016 that chronic inflammatory disease may not be cured unless the cause of airway wall remodelling is understood.
Gene linkage analyses suggested that the susceptibility to asthma or COPD is inheritable, but out of 182 candidate genes, none had been proven as a cause. Family studies showed heritability; while twin studies indicated that genetic conditions contribute less than 50% likely to develop asthma.
Recent studies on gene regulation suggest that epigenetic mechanisms may be inheritable and present as pre-conditions to develop asthma or COPD. There is evidence that the lung is sensitive during embryogenesis and early childhood to develop asthma or COPD later in life. The nature of this imprinting process seems to be epigenetic but is not well understood.
Several epigenetic mechanisms can imprint the lung for asthma or COPD, including chemical modifications of histones and DNA. These events lead to over-expression of inflammatory proteins or block anti-inflammatory proteins. Epi-genetic events are modified microRNA regulation and stability, which is associated with mitochondrial activity. The activation of epigenetic mechanisms can occur through asthma or COPD risk factors such as inhaled allergens, chemicals, cigarette smoke, and dust, as well as by physical and psychologic stress factors.
Computer models based on large data collections from the USA, Europe and Asia showed that outdoor air pollution caused 3.3 million premature deaths worldwide in 2015, which is expected to double by 2050. Indoor pollution (open fire cooking, heating) killed another 3.5 million people. Air pollution can also be caused by fine dust (<10 pm) from agriculture in less industrialised countries.
A survey by the European Union in 2016 stated that in Europe 50% of chronic inflammatory diseases are misdiagnosed and inadequately treated. Thus, more investigations into the pathogenesis of chronic inflammatory lung diseases are urgently needed if we want to reduce the burden on the patients and the society. Only if the origin of the diseases is understood, new disease-specific diagnostic tools and personalised therapies can be developed. To achieve this, investigators need access to data and tissue banks generated from human asthma and COPD lungs.
Genetic origins of chronic inflammatory lung diseases
Candidate genes for asthma susceptibility included the glucocorticoid receptors, ADAM33, TGF-β receptors, IL-33, surfactant protein D, IL-4 and interferon-λ. In COPD, the most recent candidate genes were surfactant protein-D, alpha-1-antitrypsin, hypoxia-inducible factor-1, TGF-β1 and Matrix metalloproteinase 12.
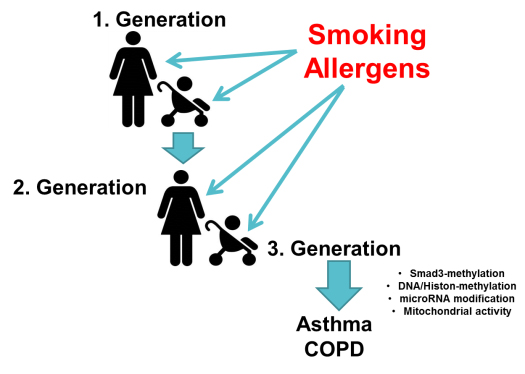
Moreover, specific gene alleles may only be relevant to the disease in Caucasians or Asians. None of the candidate genes had ever been proven to be a single cause of the diseases. There is evidence that modified gene regulation during embryogenesis imprints the lung to develop asthma or COPD later in life. Family studies performed over three generations suggested that the imprinting of the embryonal lung by cigarette smoking mothers increased the chance for their children to develop COPD by over 40%. This epigenetic imprinting of the embryonal lung was also correlated to cigarette smoking grandmothers, with the possibility of skipping one generation. Cigarette smoking lastingly altered DNA and histones methylation, thereby increasing the chance of children to develop COPD (figure 1).
Early studies analysing the effect of inhalable compounds on the structure of the airways – showed that lung structure pathologies were induced during embryogenesis. The studies showed that a second event was needed to initiate chronic lung inflammation in the offspring. In rhesus monkeys, the exposure to allergens during pregnancy and the new-born resulted in the disturbed interaction of epithelial cells with smooth muscle cells, smooth muscle hypertrophy, cytokine expression, and vascular remodelling of the airway wall. Importantly, none of these pathologies could be corrected later in life. Thus, once damaged, the lung stays damaged for the entire life. These observations suggest that epigenetics plays an important role in the pathogenesis of chronic inflammatory lung diseases.
Epigenetic events as a cause of chronic lung inflammation
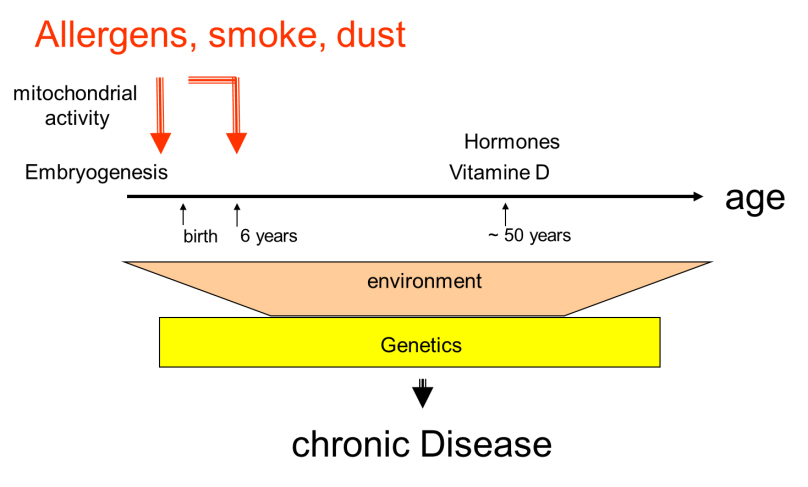
In mothers and children cohorts, the susceptibility to develop asthma was associated with the exposure of mothers and children to the same risk factors during late embryogenesis (last trimester) and early childhood (0-6 years), as summarised in figure 2.
Another risk factor to pre-dispose the lung for chronic inflammation later in life was low levels of zinc ions in mothers and children during pregnancy. This correlated with an increased risk of asthma in children (odds ratio >0.57). Zinc deficiency affects the function of a large number of proteins related to asthma, including the glucocorticoid receptors, matrix metalloproteinases and DNA methylating proteins. Furthermore, zinc finger proteins regulate hormone-dependent gene activity, RNA transcription and protein synthesis, all of which are epigenetic regulators.
Malnutrition during pregnancy increased the risk for the child to develop asthma, wheeze, and atopic diseases later in life. Risk factors for asthma can be transmitted by breastfeeding. There is also evidence for multi-generation transmission of asthma susceptibility, as mothers exposed to phthalates during pregnancy increased the risk for the next two generations to develop asthma. The exact mechanism of this trans-generational asthma susceptibility is unknown, but experimental and clinical data suggested that it is due to DNA and histone methylation and modified microRNA expression.
DNA methylation was studied in 527 children (5-12 years) born to cigarette smoking mothers and identified 20,578 methylated DNA sequences. Most of the genes were methylated at stretches of CpG repeats with unknown function.
In a second study (572 children), DNA methylation caused by cigarette smoke was linked to respiratory symptoms at the age of 3-5 years. Tobacco smoke increased gene-specific methylation by 10-fold in 26 different genes, including the aryl hydrocarbon receptor repressor (AHRR) and cytochrome P450 (CYP).
However, 50% of the affected genes encode for unknown proteins or microRNAs. In another study (245 females, 10-18 years), DNA methylation affected mainly genes encoding for Th2 cytokines, which are known to be increased during asthma. It is unknown if DNA methylation patterns in children exposed to risk factors are identical to those in adult asthma.
The importance of epithelial-mesenchymal-trophic-unit (EMTU) in lung function
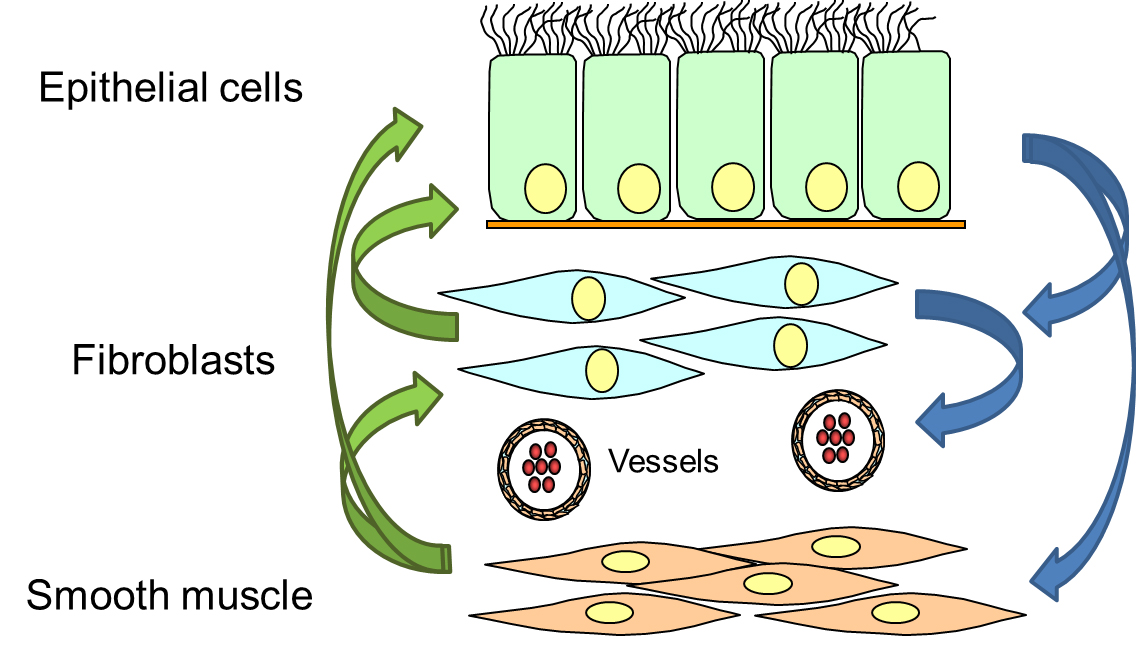
The EMTU describes the interaction of different cell types that form the bronchi. Animal models implicated that airway wall remodelling originated from over-active fibroblasts/myofibroblasts, which produced and deposed pro-inflammatory components of the extracellular matrix and proliferated faster. However, in the human lung, this pathogenesis was never fully confirmed and more recent studies on human tissues and isolated cells suggested a different mechanism.
Epithelial cells are a key regulator of the sub-epithelial mesenchymal cells. In case of epithelial injury or loss of the epithelium, the sub-epithelial cells start to proliferate, to compensate the loss of the epithelium. As a result, the thickness of the airway wall increases, leading to local hypoxia, followed by the formation of new micro-vessels and infiltration of pro-inflammatory immune cells (figure 3).
In our own asthma studies, the epithelium obtained from asthma patients controlled the function of fibroblasts and airway smooth muscle cells. Most of the available drugs for asthma and COPD can well control the secretion of pro-inflammatory cytokines, while they have no effect on proliferation or the deposition of pro-inflammatory extracellular matrix components.
Recent studies indicated that epithelial cells specifically control mitochondrial activity in sub-epithelial cells. Mitochondria can be linked to the above-mentioned epi-genetic mechanism, leading to chronic lung inflammation. However, a search by Pubmed (keywords: asthma, mitochondrial, NOT review) found only 219 publications focused on this topic. Similarly, 205 publications in COPD investigated the role of mitochondria. The inheritance of epigenetic events would be most effective if they occur in mitochondria genes, which are forwarded to the next generation only by the mother.
Malfunction of mitochondria was reported in human asthma and correlated with airway smooth muscle cell hyperplasia and increased secretion of pro-inflammatory cytokines. Epigenetic deregulation of mitochondria genes was associated with ageing, resetting gene regulation during embryogenesis, inflammation, proliferation, and cell differentiation. Interestingly, the function of mitochondrial genes or mitochondria regulating genes is controlled by microRNAs. Thus, more investigations are needed to clarify the role of mitochondria in chronic inflammatory lung diseases.
The contribution of microRNAs to chronic inflammatory lung diseases
MicroRNAs are short strands of RNA which had been regarded as useless elements for a long time. Only recently, microRNAs were recognised as essential post-transcriptional regulators of protein production. MicroRNAs act as the epi-genetic regulator of RNA-protein translation, RNA stability and mitochondria activity. Regarding asthma and COPD, 47 different microRNAs have been linked to organ malfunction and disrupted cell-cell interaction.
MicroRNAs can be detected in body fluids such as plasma and serum, which makes them candidates as new biomarkers. However, there is a lack of evidence microRNAs are specific for asthma or COPD as many have been described earlier as markers for various tumours.
However, microRNAs may present a novel form of therapy, as they are functional and stable under certain conditions in plasma and serums. As a biological drug, microRNAs can be protected by peptide envelopes and even can be taken up by cells from the bloodstream. Some on-going investigations explore the possibility to create such peptide enveloped microRNAs as a therapy for diseases where specific proteins are over-expressed.
One problem with the use of microRNAs as therapeutics is the fact that the same microRNA can be highly expressed in one cell type while being down-regulated in another cell type under disease conditions. For example, microRNA-19 is up-regulated in immune cells, while it is down-regulated in smooth muscle cells and fibroblasts of asthma patients. A similar problem was described for microRNA-21 in COPD versus lung cancer.
Further attention must be given to the fact that microRNAs have many mRNA targets. For example, microRNA-19a down-regulates the mRNA of Erk1/2 mitogen-activated protein kinase as well as the microRNA of the TGF-β receptor 1, TGF-β associated protein and tyrosine kinase. Therefore, its high expression in immune cells should result in reduced production of pro-inflammatory cytokines.
In contrast, the lack of microRNA-19a in mesenchymal cells in asthma – should lead to increased production of pro-inflammatory factors and stimulate the cell proliferation. The latter observation by our group can explain the mechanism underlying the regulation of asthmatic mesenchymal cell behaviour, which has been described by others earlier. Therefore, the use of microRNAs as therapeutic must be carefully studied to avoid unwanted side effects.
Conclusion
Lungs are vulnerable to be pre-disposed for developing chronic inflammatory lung diseases during two-time windows in late embryogenesis and early childhood. These critical phases determine the lung’s maturation, function and structure for the rest of the life.
The susceptibility to developing asthma and COPD may not be inherited through a genetic pre-condition, but rather by irreversible epigenetic events. Future studies need to investigate how epigenetic modifications become “fixed” and inheritable.
Mothers should be better informed about these risk factors and that they can do more to prevent their children from suffering lifelong malfunction of the lung.
Finally, the interaction of basic, clinical and epidemiological research is needed for better understanding the cause of asthma and COPD to find new therapies.
Please note: this is a commercial profile
Michael Roth
Research group leader pulmonary cell research
University Hospital Basel & University Basel
Tel: +41 (0)61 265 2227
https://biomedizin.unibas.ch/nc/about-us/people/profil/profile/person/roth-3








")